


Motor Neurone Disease (MND)
Diagnosis, differential diagnosis, management and complications of MND
Overview
- Motor neurone disease is a progressive, degenerative disease affecting exclusively motor nerves.
- It is a life-limiting condition, and carries a poor prognosis making it an important diagnosis to consider, make, understand and explain
- It is also vital not to misdiagnose a treatable or reversible mimic of MND
- Patients are often diagnosed late as a consequence and can present in the very late stages of the disease
- Management is centred on supporting weak muscles of breathing, ensuring adequate safe nutrition, managing secretions to minimise life-threatening aspiration, and good palliative care
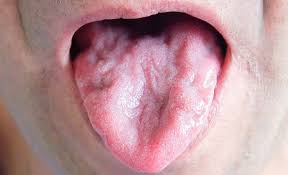
Tongue wasting in MND
Epidemiology
- The approximate incidence is 2.6/100,000 per year (UK), with a prevalence of 5-7/100,000 population. It affects men slightly more than women, with lifetime risks of 2.1/1000 in women and 2.9/1000 in men
- Both sexes show peak incidence at age 75-79
Causes
- The cause of motor neurone disease is unknown
- There are examples of familial MND (involving mutations for SOD 1 and c9ORF amongst others) but the majority is sporadic
Presentations of motor neurone disease
- The classic picture (ie. exam case) for MND is “an exclusive motor deficit with mixed upper and lower motor neurone signs”, usually with evidence of wasting and fasiculations
- As the name implies this is a degenerative disease selectively affecting motor nerve nerves. It therefore presents with weakness, the clinical picture depending on which part of the neuraxis is affected first.
- Patients may fit into one of four subdivisions of presentation, although usually have features which overlap as the disease progresses
- The way to think of these is whether it predominantly cord or brainstem, and whether the picture is more UMN or LMN at the beginning)
Amyotrophic lateral sclerosis (cord> brainstem, UMN > LMN)
- Predominant loss of corticospinal tract, causing a progressive, painless, spastic paraparesis. There will be both UMN in form of increased tone and reflexes, and LMN in terms of wasting and fasiculations
- This is the most common form of motor neurone disease
- In the USA this is the term used for all MND
- Differential diagnosis: spinal cord pathology eg tumour, cervical spondylosis
Progressive muscular atrophy (cord >brainstem, LMN > UMN)
- Prominent wasting, weakness and fasiculations, often small muscles of the hand affected first
- Mixed UMN and LMN signs with either decreased or increased reflexes
- Differential diagnosis: Lead neuropathy, myasthenia, multifocal motor neuropathy, diabetic amyotrophy, polio/post polio (ideally include links on each of these as hyperlinks which will have a box of info when you hover over them- will put a box in!)
Progressive bulbar palsy (brainstem> cord, LMN > UMN)
- NB. But can be prominent UMN signs with spastic palate and tongue involvement causing a “pseudobulbar palsy” which just means UMN bulbar problems.
- The pathology lies predominantly in the cranial nerve nuclei
- Progressive weakness of bulbar muscles will lead to problems with speech, swallowing (think secretion management and aspiration risk), and earlier involvement of breathing with diaphragmatic weakness
- Always ask about shortness of breath, particularly on lying flat
- Patients often describe nasal regurgitation.
- More common in women
Primary lateral sclerosis (brainstem/cord, UMN exclusively)
- Rare form
- Progressive tetraparesis and pseudobulbar palsy
Video of fasciculations in motor neurone disease
Differential diagnosis of MND
- Key differentials are in bold. The examination/investigation which is most helpful in identifying the mimic is below the differential diagnosis
- Cervical radiculopathy
- MRI spine shows multi-level disc compression
- Syringomyelia
- MRI spine shows syrinx (expansion of the CSF space in the centre of the cord)
- Syphilic pachymeningitis
- Now very rare, untreated syphilis used to be much more common, and can cause a patchy meningitis, leading to poor function of the exiting nerve roots in the brainstem ad spinal cord
- Test for EIA/VDRL serology
- Motor neuropathy
- Lead/heavy metal poisoning is the most common cause of a progressive motor neuropathy – check serum levels
- Multifocal Motor Neuropathy is a rare inflammatory condition involving antibodies to nerve root antigen – check “anti ganglioside antibodies”; MMN has positive anti GM1 antibodies
- Also nerve conduction will be different to MND – and show conduction block although this can be difficult to identify
- Spinal muscular atrophies
- Rare genetic disorders causing progressive muscle atrophy, of which there are a range that vary in severity, age of onset and rate if progression. Genetic tests are available
- Kennedy’s Syndrome “Spinal and bulbar muscular atrophy”
- X-linked condition, causing spinal and bulbar muscular atrophy. Only LMN pathology. Pathology is due to a mutation in the androgen receptor and therefore also has endocrine manifestations of androgen insensitivit
- Usually presents in young-middle aged adult males
- Ask about muscle cramps. Look for chin fasciculations and gynaecomastia
Diagnosis and investigations in MND
- It is critical to diagnose MND accurately; make sure you are certain before counselling a patient and their loved ones about their prognosis
- EMG/NCS is the most helpful investigation
- Looking for evidence of (usually wide spread) denervation
- The typical finding is chronic partial denervation with preserved motor conduction velocity
- NB Primary lateral sclerosis will not show denervation as it is exclusively UMN
Staging
- There is no formal staging system for MND. However, it often progresses quicker than anticipated and it is important to plan ahead for inevitable deterioration
- Bulbar presentations are generally associated with a poorer prognosis, presumably because respiratory muscles are involved earlier, because feeding is limited (motor neurone disease causing significant wasting and weight loss even without this) and due to the risks associated with aspiration
- Death is certain, usually within a year to maximum five years after diagnosis
Initial management
- Communication
- Once the diagnosis is definite the first priority is in communicating this with the patient in a sensitive manner
- Multidisciplinary team
- In all chronic conditions but particularly MND, getting patients linked in to the appropriate members of the multidisciplinary team is vital
- Patients will vary in their stage of disease at presentation and also their rate of decline, but it is better to get the entire MDT involved as soon as the diagnosis is established and discuss the need for nutritional and respiratory support early
- Riluzole
- Riluzole is the only pharmacological treatment licenced for use in MND. It has been shown to increase average survival by approximately 2-3 months
- A Cochrane review found a 9% increased chance of surviving a year. Its mode of action is controversial – it preferentially blocks TTX sodium channels but its effects are thought to be due to anti-glutamatergic action, by increasing glutamate uptake, accelerating glutamate clearance and preventing pre-synaptic glutamate release
Further management
Nutrition
- This is vital as motor neurone disease is catabolic – patients have often lost huge amounts of weight in the run up to their diagnosis, even without bulbar problems limiting their intake
- Bulbar problems require gastrostomy (always in discussion with the patient and accepting their preference for risk feeding if expressed). There are two ways of placing this:
- PEG (percutaneous gastrostomy) – fitted via OGD (endoscopic placement). The stomach is visualised through the oesophagus and then a stoma is formed from the stomach to the outside world. This requires the patient to be able to lie flat which may be problematic if they have respiratory muscle weakness
- RIG (radiologically inserted gastrostomy) – placed under radiological guidance but usually requires the patient to have a BMI > 17
Venilatory Support
- Respiratory weakness requires ventilatory support
- Early discussion about patient’s preferences are important as some patients do not want to use non invasive ventilation
- It is a life-prolonging treatment, which can initially be used just at night but which patients will become increasingly dependent on, and works by providing pressure to promote air flow in and out of the lungs allowing them to function as normal
- Intubation is considered inappropriate in all but extreme circumstances as patients are very difficult or impossible to wean off ventilators due to their respiratory muscle weakness, and have no potential for reversal of their condition
- Discussions around ceiling of care should be had early, be clearly documented and easily available
Complications of MND
- Complications come from weakness in important muscles groups
- Respiratory failure
- Respiratory muscle involvement causes type II respiratory failure, with symptoms of breathlessness on exertion or when lying flat
- Examination: poor inspiratory effort, lack of abdominal movement with respiration, and even the bounding pulse of CO2 retention
- Investigations: Gradually climbing carbon dioxide levels on arterial blood gas
- CXR showing raised hemi-diaphragm
- EMG of the intercostal or diaphragm showing denervation
- The only treatment is to provide ventilation support in the form of non-invasive ventilation
- Aspiration pneumonia
- Bulbar muscle weakness means that patients do not manage to swallow their secretions putting them at high risk of aspiration pneumonia
- Often this is chronic and is called “silent aspiration” meaning patients breathe in very small volumes of secretion over a long period of time. Eventually this will lead to a lower respiratory tract infection which is difficult to compensate for with weak respiratory muscles
- N.B. Pneumonia will cause a predominantly type 1 respiratory failure (with hypoxia) although in the presence of respiratory muscle weakness expect the CO2 to be higher than you would expect from their elevated respiratory rate if breathless
Prognosis of MND
- Motor neurone disease is fatal but the progression is variable
- The best indicator is how quickly a person is diagnosed – the longer this takes, the longer they are likely to survive
- It is rare to survive beyond five years from diagnosis
Video overview of motor neurone disease
Famous people with MND
- Stephen Hawking
- This is not the typical picture of motor neurone disease and there are plenty of theories around why he has survived so much longer than expected. ‘The Theory of Everything’, written from his wife Jane’s memoirs is a beautiful portrayal of the destructive course of motor neurone disease.
- Lou Gehrig
- In the USA, MND is often called ‘Lou Gehrig’s disease’ after a famous baseball player who died very young from amyotrophic lateral sclerosis.
Now click here for a full list of MND differential diagnoses
Targeted list of motor neurone disease mimics
… Or click here to learn how to diagnose and manage myasthenia gravis
Click the tags below for similar content: