


Definition and pathogenesis of myelofibrosis
- Myelofibrosis is one of the myeloproliferative neoplasms, which may arise de novo or progress from polycythaemia vera or essential thrombocythaemia.
- The malignant clone of myeloid progenitors causes an increase in aberrant megakaryocytes, and a hypercellular bone marrow with increased white cells and platelets.
- These megakaryocytes promote bone marrow fibrosis, by secreting growth factors
- Progressive marrow fibrosis inhibits haematopoiesis causing low cell counts, so extramedullary [outside bone marrow] haematopoietic tissue develops, particularly in the spleen
- Over 50% have mutations in JAK2, CALR or MPL
Epidemiology of myelofibrosis
- Myelofibrosis is one of the rarer myeloproliferative diseases, with a UK incidence of 0.5 per 100,000 per year
- Slight male preponderance
- Disease of the elderly, median age 73 at diagnosis.
Clinical features of myelofibrosis
- Early presentations
- Incidental finding of raised platelet count or white cell count
- Thrombosis at unusual sites (e.g. abdominal veins)
- Recurrent gout
- Late presentations (more commonly)
- Splenomegaly (may cause discomfort, early satiety, or an incidental examination finding)
- Hepatomegaly
- Anaemia
- Constitutional symptoms: sweats, weight loss, fatigue, fevers, bone pains
Investigation of myelofibrosis
- Common blood tests: FBC and blood film (fig.1 and 2), U+E, LFT, Uric acid, LDH
- Genetic testing for JAK2 V617F mutation, and mutations in CALR and
- Ultrasound abdomen to assess for splenomegaly
- Bone marrow aspirate and trephine (note aspirate is often impossible ‘dry tap’)
Figure 1. Blood film showing tear drop red cells (arrowheads), nucleated red blood cells (filled arrow) and immature granulocyte precursors (hollow arrows).
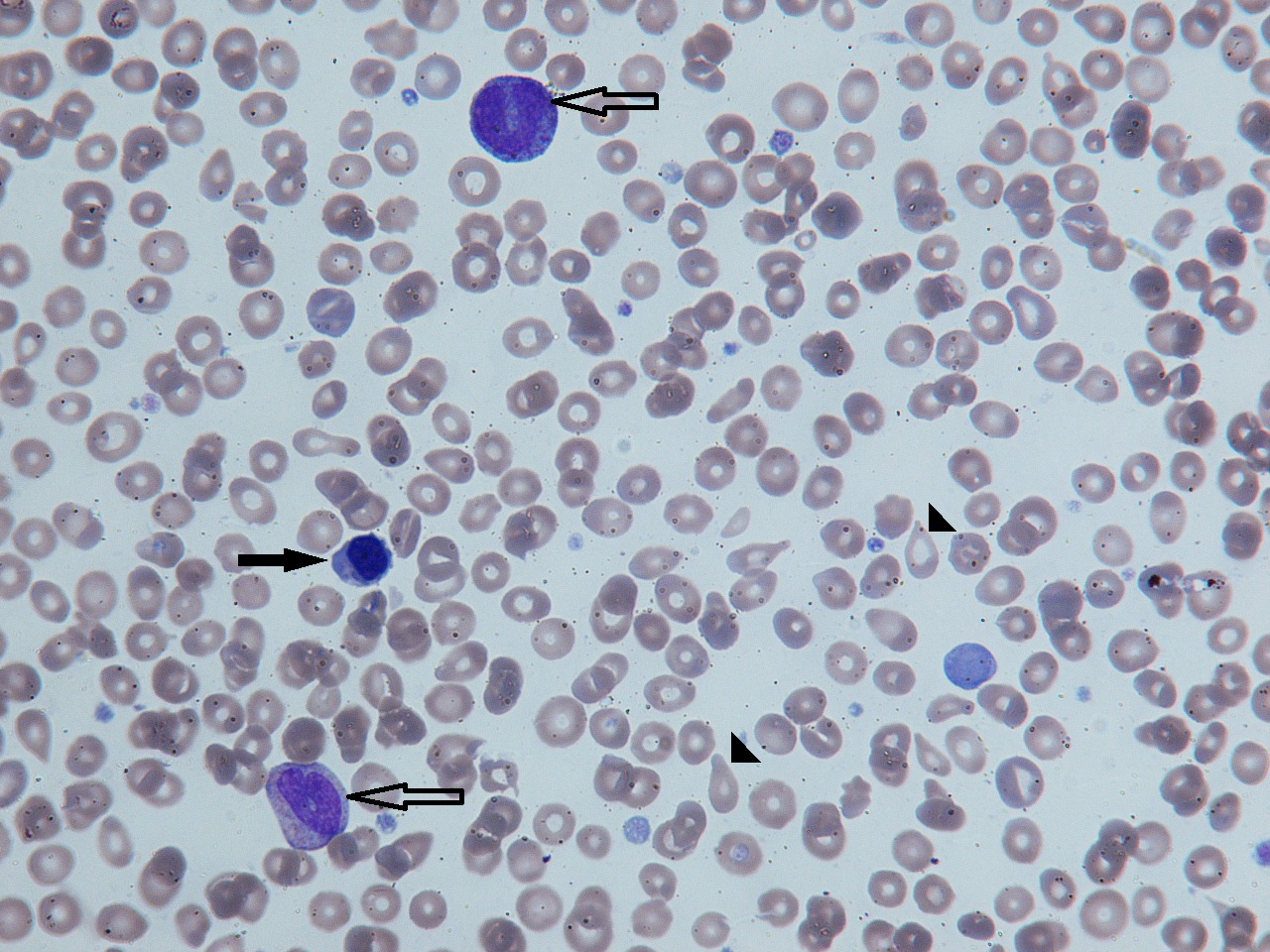
Figure 2. In advanced disease circulating blasts may be seen (hollow arrows)

Diagnostic criteria for myelofibrosis
- World Health Organisation (WHO) criteria and British Committee for Standards in Haematology (BCSH) criteria largely agree, though the former includes a category of ‘prefibrotic myelofibrosis’
- BCSH criteria require:
- Fibrosis on bone marrow trephine, scored at 3 or 4 out of 4, and
- A culprit mutation in JAK2, CALR or MPL, or the absence of any reactive cause of fibrosis, and
- Any two of
- anaemia
- leukoerythroblastic blood film
- tear-drops red cells on blood film
- constitutional symptoms
- palpable splenomegaly
- other evidence of extramedullary haematopoiesis
Complications of myelofibrosis
- Splenic vein thrombosis
- Portal hypertension
- Bone marrow failure
- Progression to acute myeloid leukaemia
Differential diagnosis of myelofibrosis
- Other myeloproliferative disease
- Essential thrombocythaemia
- Polycythaemia rubra vera
- Chronic myeloid leukaemia
- Other haematological causes of marrow fibrosis
- Hairy cell leukaemia
- Metastatic non-haematological cancer
- Rarely, long-term use of thrombopoietin mimetic drugs (eltrombopag, romiplostim)
Prognosis of myelofibrosis
- The Dynamic International Prognosis Scoring system can be used to estimate prognosis based on age, constitutional symptoms, white count, haemoglobin and peripheral blood blast count
- Score 0: low risk (median survival likely >18 years from diagnosis)
- Score 1-2: intermediate-1 (median survival 14.1 years from diagnosis)
- Score 3-4: intermediate-2 (median survival 4 years)
- Score 5: high risk (median survival 1.5 years)
Management of myelofibrosis
- For DIPPS low risk and asymptomatic intermediate-1 cases, active monitoring
- For intermediate-2 or high risk, if transplant-eligible, an allogeneic stem cell transplant is the only curative option
- In other patients, treatment is symptom-driven:
- for symptomatic splenomegaly or constitutional symptoms, the JAK2 inhibitor ruxolitinib is used. Other options include interferon-alpha and hydroxycarbamide
- for anaemia, erythropoietin should be used if serum levels are low, or danazol if levels are replete. Transfusion may be required for refractory anaemia.
- other cytopenias may improve with thalidomide plus prednisolone
Passamonti et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010:115:1703-1708.
Questions about myelofibrosis
- How can early myelofibrosis be distinguished from essential thrombocytosis?
- There may be clinical features, such as weight loss or splenomegaly, which make myelofibrosis much more likely. If not, then bone marrow trephine findings are key to make the distinction.
- Does ruxolitinib improve survival in myelofibrosis?
- Ongoing follow up data from the COMFORT trials has now shown an improvement in overall survival in those groups allocated to ruxolitinib (American Society of Haematology annual meeting abstracts, 2016). However these trials were confined to patients with symptomatic splenomegaly or constitutional symptoms, and it is not known whether ruxolitinib improves survival in asymptomatic patients.